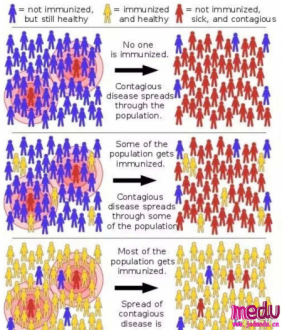
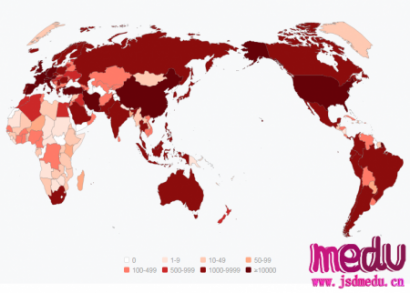
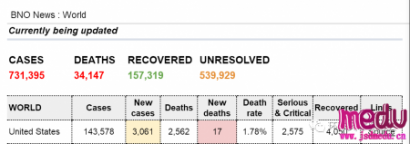
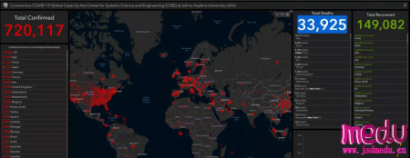
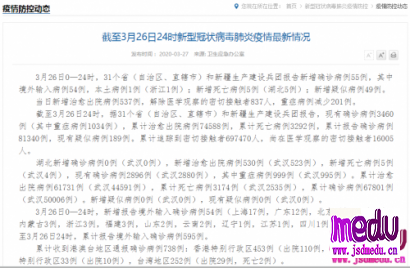
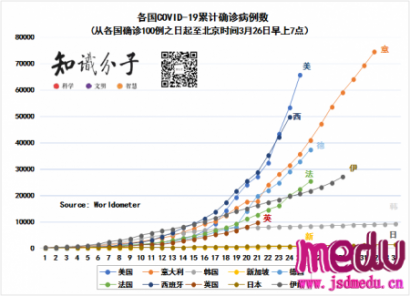
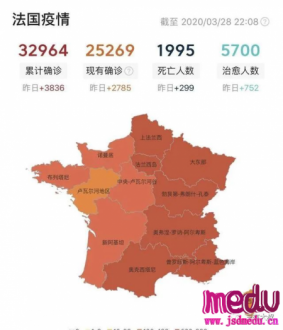
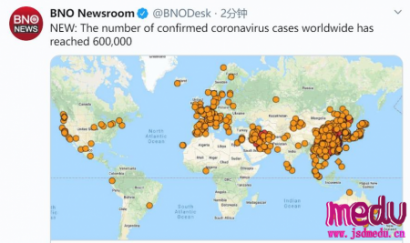
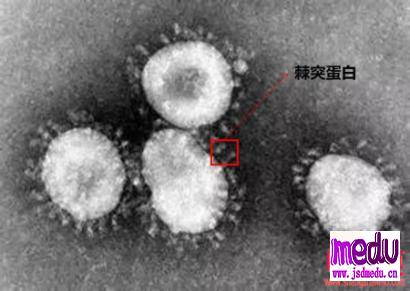
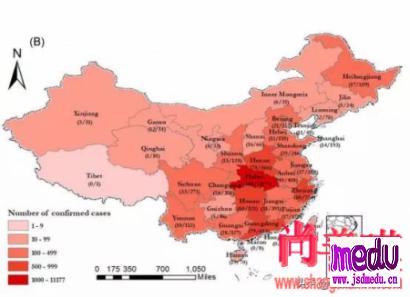
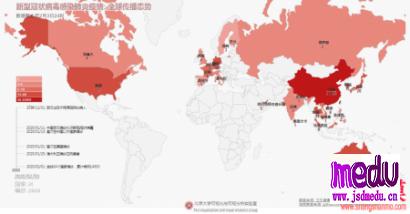
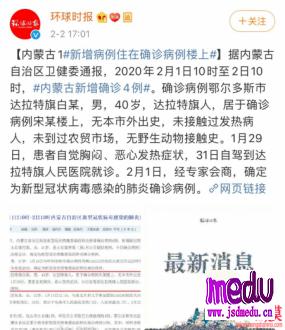
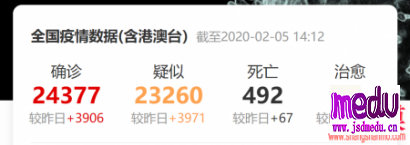
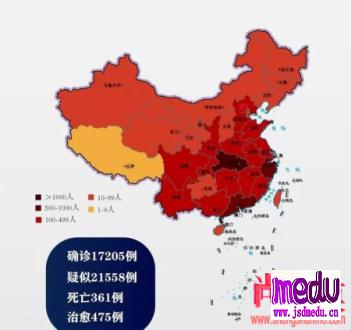
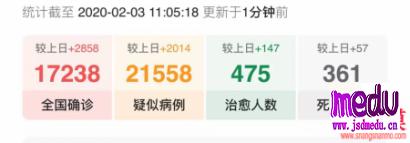
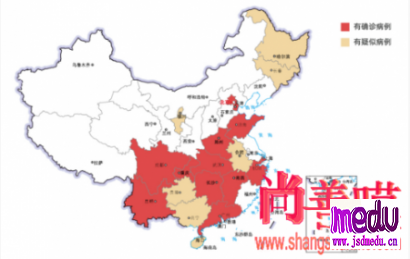
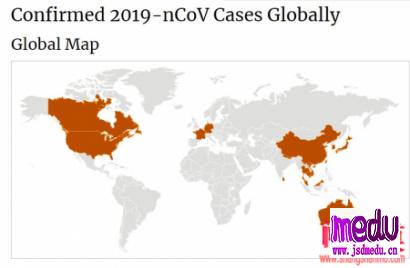
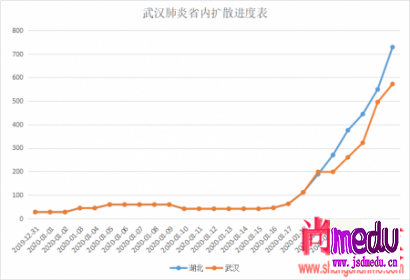
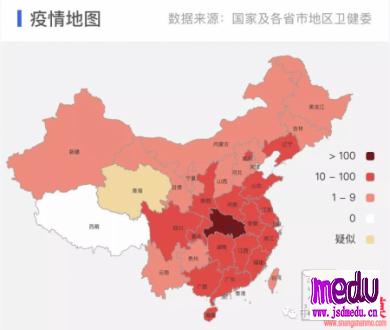
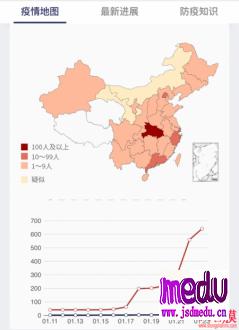
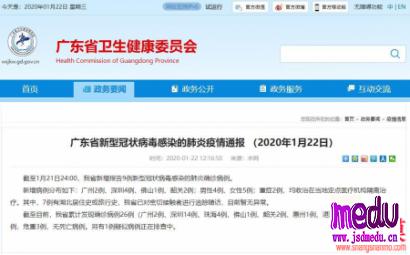
连日来,新型冠状病毒引起的肺炎疫情发展牵动人心。
相关的研究工作也在开足马力抓紧时间进行。
包括病毒核酸检测,病毒基因组测序等。
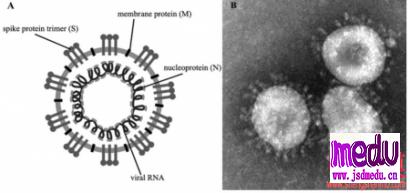
关于冠状病毒
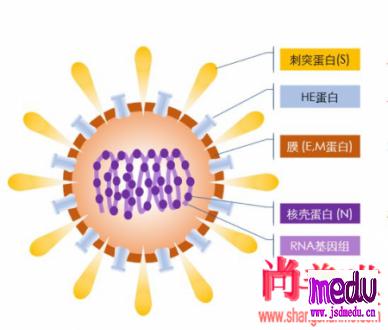
冠状病毒属于套式病毒目、冠状病毒科、冠状病毒属,是一类具有囊膜、基因组为线性单股正链的RNA病毒,是自然界广泛存在的一大类病毒。
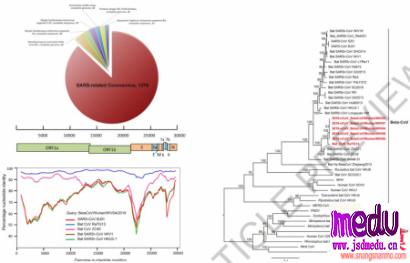
某些冠状病毒会感染人类并引起疾病,2019年12月在首先发现的冠状病毒是一种先前尚未在人类中发现的病毒株系。
2020年2月11日,该病毒被国际病毒分类委员会(theInternationalCommitteeonTaxonomyofViruses)冠状病毒研究小组(CoronavirusStudyGroup,CSG)命名为“SARS-CoV-2”(SevereAcuteRespiratorySyndromeCoronavirus2),同时,由该病毒感染引起的疾病被WHO命名为“COVID-19”(CoronaVirusDisease2019)。
——国家基因组科学数据中心(NGDC)
相对于DNA病毒,RNA病毒往往具有更频繁的变异。
病毒复制和竞争在每一个受感染的宿主体内产生一个复杂的变种混合物。
群体内所有变异的解析对于理解进化、准种动力学(QuasispeciesDynamics)、耐药性和免疫逃逸至关重要。
如何区分整个基因组中可能只有少数SNV不同的变体来进行深入研究?必然需要依赖于更准确的基因组碱基序列信息。
而在满足高精度要求的同时,只有PacBioSMRT测序技术能提供的更长读长范围。

HiFiReads生成原理示意图
随着近几年PacBio测序技术的不断提升,SMRT测序技术的酶读长度增加到了平均100kb,最长的酶读长更是超过300kb。
在CCS(CircularConsensusSequencing)环形一致性的测序模式下,可获得兼顾长读长与高度准确性的HiFireads。
相对于读长不过几百bp的NGS,PacBioSMRT测序在提供准确碱基序列的同时,能够跨越更长的片段范围,为病毒基因组中复杂的突变提供更为完整的信息。
SMRT测序是目前真正的高准确率且长度长测序技术,优势远超NGS或者其他长度长技术以及他们的结合。
基于这一方法,研究人员能够对病毒的基因组进行更加准确的研究!

接下来,向大家分享一篇运用超高深度的SMRT测序技术进行HIV病毒变异研究的文章。

由于艾滋病病毒HIV属于逆转录病毒,抗逆转录酶药物成为了艾滋病治疗的重要手段。
然而在联合抗逆转录酶药物治疗(cART)时,一些艾滋病患者依然会出现较为严重的神经系统损伤,这有可能是由于在病人脑组织中,HIV的持续感染及进化选择所致。
为了验证这一假设,文章作者对一个HIV+/cART+受试者的七个尸检组织进行取样,其中包括了三个大脑和四个非大脑部位,通过核酸提取,巢式PCR对env基因进行全长扩增,SMRT建库,以及在PacBioRSII平台上进行测序,获得了5万条高质量的全长HIV包膜基因的序列,其序列长度可达2200bp。

对高质量共有序列进行比较分析,构建系统发育树。
分析表明,脑源性病毒呈现明显的聚类,并且在进化上不同于脑外组织中的病毒。
作者在文中也明确地表明,所研究的三个脑组织中的每个病毒在遗传上都是截然不同的,而来自所有外围组织的变体则遍布整个发育树,并且不依附于脑部序列的进化分支。
对于这样的分析,作者也在文中指出,不足300bp的NGSreads,必然需要重新进行组装。
但HIV病毒的高度变异,则大大提升了组装的不确定性。
然而借助于PacBio测序技术的长读长优势,这一分析能够通过2200bp的全长序列,分析整个包膜基因的变化,从而给出更为准确的判断。
目前,SMRT测序针对高度变异的RNA病毒进行的研究已经发表了多篇文章,如:
案例1:用SMRT测序技术进行急性MERS-CoV感染患者血清中的刺突基因缺失的准种测定
SpikegenedeletionquasispeciesinserumofpatientwithacuteMERS‐CoVinfection
通过发现一名患者的血清中冠状病毒刺突基因中有530bp片段的缺失,确认了人类感染了一种以准种形式存在的新型MERS-CoV基因变异。
并推测了其可能的起源以及对预测的峰值蛋白功能的影响。
https://onlinelibrary.wiley.com/doi/full/10.1002/jmv.24652
案例2:分析HIV病毒准种体系
SequencingComplexMixturesofHIV-1GenomeswithSingle-BaseResolution
埃默里大学(EmoryUniversity)AIDS研究中心与赞比亚的研究组进行合作,研究HIV-1病毒在传染的过程中准种的变化。
以往他们采用的是单基因组扩增测序技术(SGA),利用该方法,在8-10天内只能完成30个病毒基因组的分析。
而利用PacBio单分子测序研究者在不到90分钟的时间即可完成1000-3000个病毒基因组的分析工作。
https://www.pacb.com/proceedings/sequencing-complex-mixtures-of-hiv-1-genomes-with-single-base-resolution/
案例3:SMRT分析用药后HIV病毒准种变化
Antibody10-1074suppressesviremiainHIV-1-infectedindividuals
SMRT测序分析第0天和注射第四周1HD1这个病人病毒进化情况。
通过对比第4天与第0天,发现巨大的变化差异。
https://www.nature.com/articles/nm.4268
案例4:SMRT测序分析HIV-1耐药性突变(HDRM)
HIV-1耐药性突变(HDRM)的发展是抗逆转录病毒疗法临床失败的主要原因之一。
通过根据患者的HDRM资料采用个性化的抗HIV方案,可以提高治疗成功率。
相比Sanger与NGS,PacBio的长读长测序技术提供的准确可靠序列用于HIV准种级HDRM分析。
对患者艾滋病毒样本的案例研究表明,使用PacBio方法生成的准种视图提供了更高的检测灵敏度,并且对于理解HDRM情况更为全面。
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4775093/
回到此次疫情的病原体SARS-CoV-2,该冠状病毒拥有着RNA病毒中少见的大的基因组,其长度接近30kb! 目前PacBioSMRT测序CCS模式所获得的HiFireads,可对病毒进行单分子准确度高达99%的测序,其片段长度已经可达20kb,并且这一数字还会在进一步的研发后有更大提升。
科技工作者可以利用该突破技术对每一个受感染的宿主体内的复杂的变种混合物进行更有价值的深度分析。
为病原学研究、诊断产品及技术、药物研究、疫苗研究等多方面应用的相关研究提供更坚实的基础。
目前,已经有很多病毒研究机构基于PacBio测序技术开展了相关的研究,您也可以通过以下链接,了解更多有关PacBio测序技术在病毒研究方面的内容。
不明原因的病毒?
PacBioSMRT测序+单细胞转录组测序深度解析流感病毒感染的细胞
贝瑞基因推出高灵敏度三代测序试剂盒阻击新冠病毒变异
文末,向奋斗在一线的医务工作者、科研工作者、各社区奋斗的一线人员们致以最崇高的敬意!